Huntington’s Disease: A Genetic Neurodegenerative Disorder of Movement, Mind, and Inheritance
Huntington’s disease (HD) is a progressive, inherited neurodegenerative condition marked by a triad of movement disturbances, cognitive degeneration, and psychiatric symptoms. It’s caused by an autosomal dominant mutation in the HTT gene on chromosome 4 and remains a powerful model for understanding the links between genes, brain function, and behavior.
1. Historical Background and Eponym
George Huntington (1850–1916) was an American physician born in East Hampton, Long Island, into a three-generation family of doctors .
Drawing upon his father’s and grandfather’s case histories, in February 1872, at just 22 years old, he delivered a landmark lecture “On Chorea” and published it soon after .
He noted three signature features:
Adult-onset chorea
Hereditary transmission, never skipping a generation
Gradual onset of dementia and suicide risk
In William Osler’s words: “a disease more accurately, graphically or briefly described” .
Huntington chose to remain a dedicated family physician, believing he could best serve patients outside academia .
2. Genetic Cause and Inheritance
The disease results from a CAG trinucleotide repeat expansion in the HTT gene (aka IT15):
Chromosome: 4p16.3
Normal allele: ≤ 26 repeats
Intermediate: 27–35 (no symptoms)
Reduced penetrance: 36–39
Full penetrance: ≥ 40 repeats
This mutation leads to polyglutamine-expanded huntingtin protein, which disrupts neuronal function. Anticipation—earlier onset in successive generations, especially via paternal inheritance—is common .
3. Pathophysiology
Mutant huntingtin undergoes misfolding and aggregation, especially in the striatum (caudate and putamen), disrupting the basal ganglia circuits that regulate movement, cognition, and emotion .
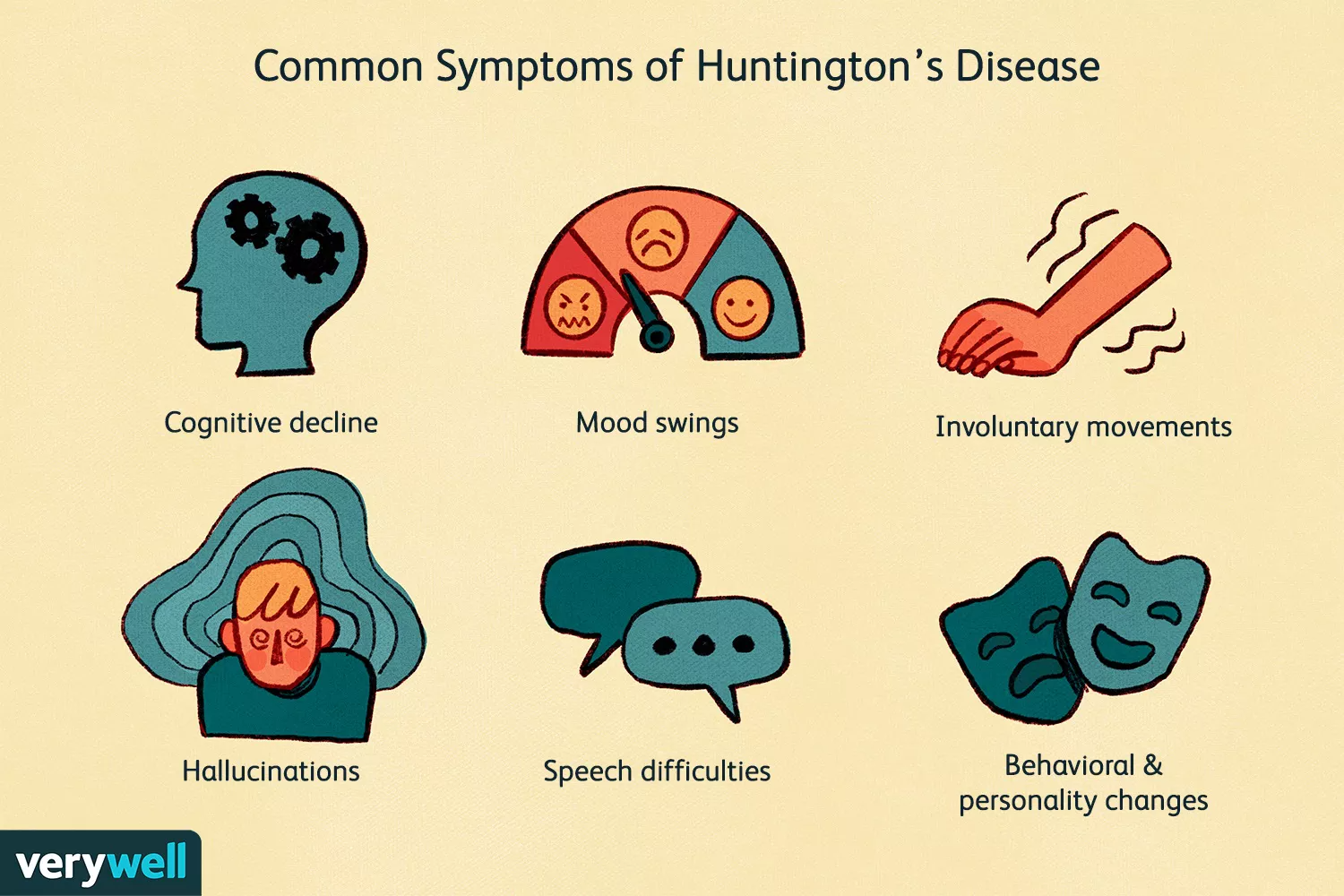
4. Clinical Presentation
Symptoms usually emerge during mid‑adulthood (30–50 years), though juvenile forms exist (< 20 years).
Motor Symptoms
Chorea: involuntary, dance‑like movements
Bradykinesia, rigidity, dystonia
Ataxia, falls, dysarthria, dysphagia
Cognitive Decline
Executive dysfunction (decision‑making, planning)
Memory impairment, slowed processing
Progressive dementia
Psychiatric Manifestations
Depression, irritability, anxiety, apathy
Obsessive‑compulsive traits, psychosis in later stages
5. Diagnosis
Diagnosis combines:
Family history and clinical signs
Genetic testing detecting CAG expansion in HTT
Brain imaging: MRI/CT may show caudate atrophy and enlarged ventricles
Predictive testing is available but must be accompanied by pre- and post-test genetic counseling due to its profound implications .
6. Management
There is no cure, but symptomatic care can improve life quality:
Medications
Tetrabenazine, deutetrabenazine: reduce chorea
Antipsychotics: address psychiatric and movement issues
Antidepressants and mood stabilizers
Supportive Therapies
Physical/occupational therapy for mobility
Speech therapy and nutritional support for swallowing issues
Psychological support, family/caregiver counseling
Advance care planning is essential due to progressive decline.
7. Research and Emerging Treatments
Current research focuses on:
Gene silencing (e.g., antisense oligonucleotides)
Experimental CRISPR/Cas9 editing
Stem cell approaches
Neuroprotective agents, targeting protein aggregation and mitochondrial function
Trials are ongoing worldwide.
8. Summary Table: Huntington’s Disease Snapshot
9. Conclusion
Huntington’s disease is one of the earliest and most illustrative examples of genetic neurodegeneration, clearly linking a gene mutation to brain function and clinical outcome. George Huntington’s compassionate observation over family generations sparked a century of scientific progress.
Although currently incurable, symptomatic therapies and advancing gene-based treatments offer hope. Early diagnosis, comprehensive care, and research into the disease’s molecular underpinnings empower patients, families, and clinicians in preparing for its challenges — and one day, overcoming them.
Comment